

Proteins are frequently called the building blocks of life because they are found literally everywhere: in our cells, bones, muscles, hormones, and enzymes. They are involved in every activity of biological existence. It is thanks to proteins that our body has structure. They help carry the oxygen that reddens our blood; in the form of enzymes they digest our food, synthesize essential substances, and break down waste products; and the chromosomes which pass on our characteristics to our children include protein in their structure. Their importance to all biological life cannot be understated.
Over the past 20 years, we have witnessed remarkable progress in improving our understanding of the human genome, driven by rapid advances in the speed and accuracy of gene sequencing. Today’s high throughput genomic sequencers are a far cry from the ones used on the Human Genome project, which took the best part of 15 years to complete. Today, researchers can have the necessary sequencing done in hours at a cost of $1000.
By contrast, we are yet to see such dramatic disruption take place in the adjacent field of the proteome. The proteome is the complete set of proteins that is – or can be – expressed by a genome, or an organism. In contrast to the human genome, which has recently been mapped out in its entirety, at present, there are still disagreements among researchers as to how many proteins actually exist. Many of the ones we think exist, we have only predicted. The Human Proteome Project (HPP) is attempting to collate and analyse all the data and create a database of all human proteins. The latest version of this database has around 20,000 predicted, of which 16,518 are supported by solid evidence (“confident protein identifications”). Only about one-third of those have had their 3D structures determined experimentally, and in many cases, those structures are only partially known. Having a clear record of all the possible proteins will allow us to improve our understanding of how proteins interact with each other, and the therapeutic targets in an event when a protein is not behaving as it should.
Part of the reason the proteome is such a tough complex to analyse is the way proteins take shape. To get a bit more technical, a protein is a complex structure that consists of either just a few or up to thousands of amino acids. Once the amino acids are in place, the protein “folds” into a complex 3-dimensional shape. Its shape in part dictates how it interacts with other proteins.
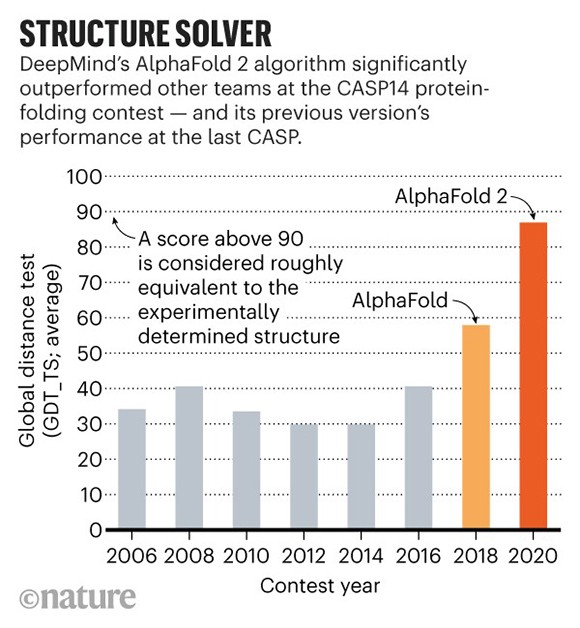
Accurately modelling these shapes and interactions is fiendishly complex. Naturally, researchers have been trying to enlist the aid of computer modelling tools to help them predict protein shapes. Every two years, a competition called CASP (Critical Assessment of Structure Prediction) is held to try and accurately model the shape of proteins based on their amino acid structure alone. Computer-software entries are judged against structures of the same proteins determined using experimental methods.
In a stunning leap forward, in 2020, a novel artificial intelligence tool called AlphaFold, developed by Google’s subsidiary DeepMind in London, stunned the scientific community by dominating the CASP competition (see chart above).
Since then, it has gone on to predict the structure of nearly the entire human proteome, along with almost complete proteomes for various other organisms, ranging from mice and maize to the malaria parasite. This year, DeepMind plans to release a total of more than 100 million structure predictions. That is half of all known proteins. Importantly, freely available open-source code for AlphaFold needed for specialists to run their own versions of the tool has now been published, igniting researchers’ interest in the space.
Researchers say the resource has the potential to revolutionize the life sciences. Understanding and correcting improperly folded proteins can hold the key to developing effective treatments of many severe diseases.
Having witnessed a revolution in genetics over the past 20 years, driven by innovative hardware, are we about to see another one in proteome analysis, driven this time by cutting edge software?